

 Print | Share
Print | Share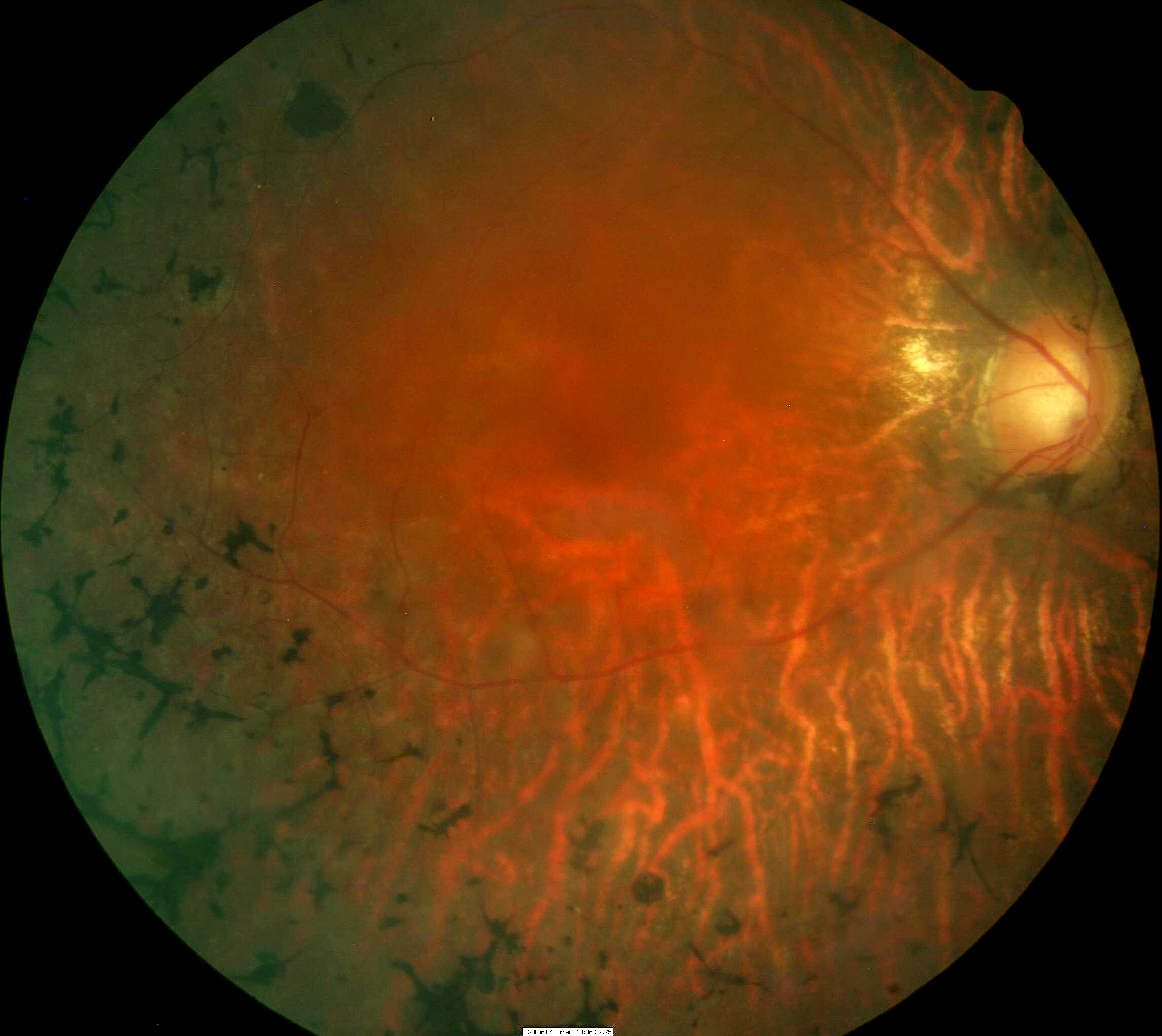
Midperipheral retinal hyperpigmentation often
seen in retinitis pigmentosa
ICD-10 Diagnosis Code:
H35.52–Pigmentary retinal dystrophy
Title
Retinitis Pigmentosa
Category
Other Retinal Disorders
Description
Retinitis pigmentosa is a group of genetic diseases that causes retinal degeneration and severe visual impairment.
Retinitis pigmentosa (RP) is a family of retinal dystrophies and retinal pigment epithelium dystrophies caused by molecular defects in multiple genes. The name “retinitis pigmentosa” is inaccurate, because the word retinitis suggests an inflammatory condition, and inflammation has not been found to be a dominant feature of this condition.
 |
Although the clinical findings in retinitis pigmentosa vary from patient to patient, the disease is characterized by the following retinal findings:
|
|
 |
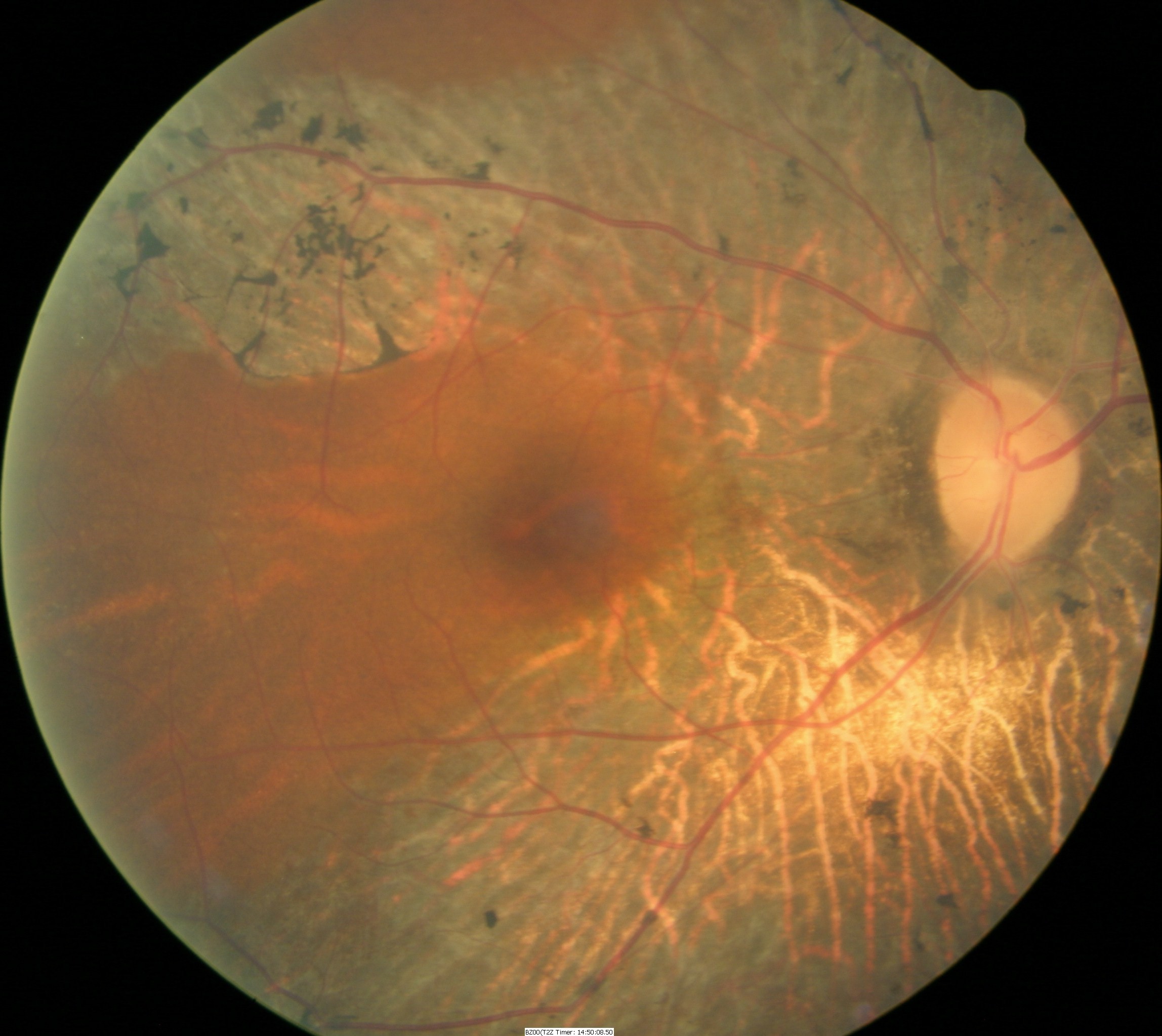
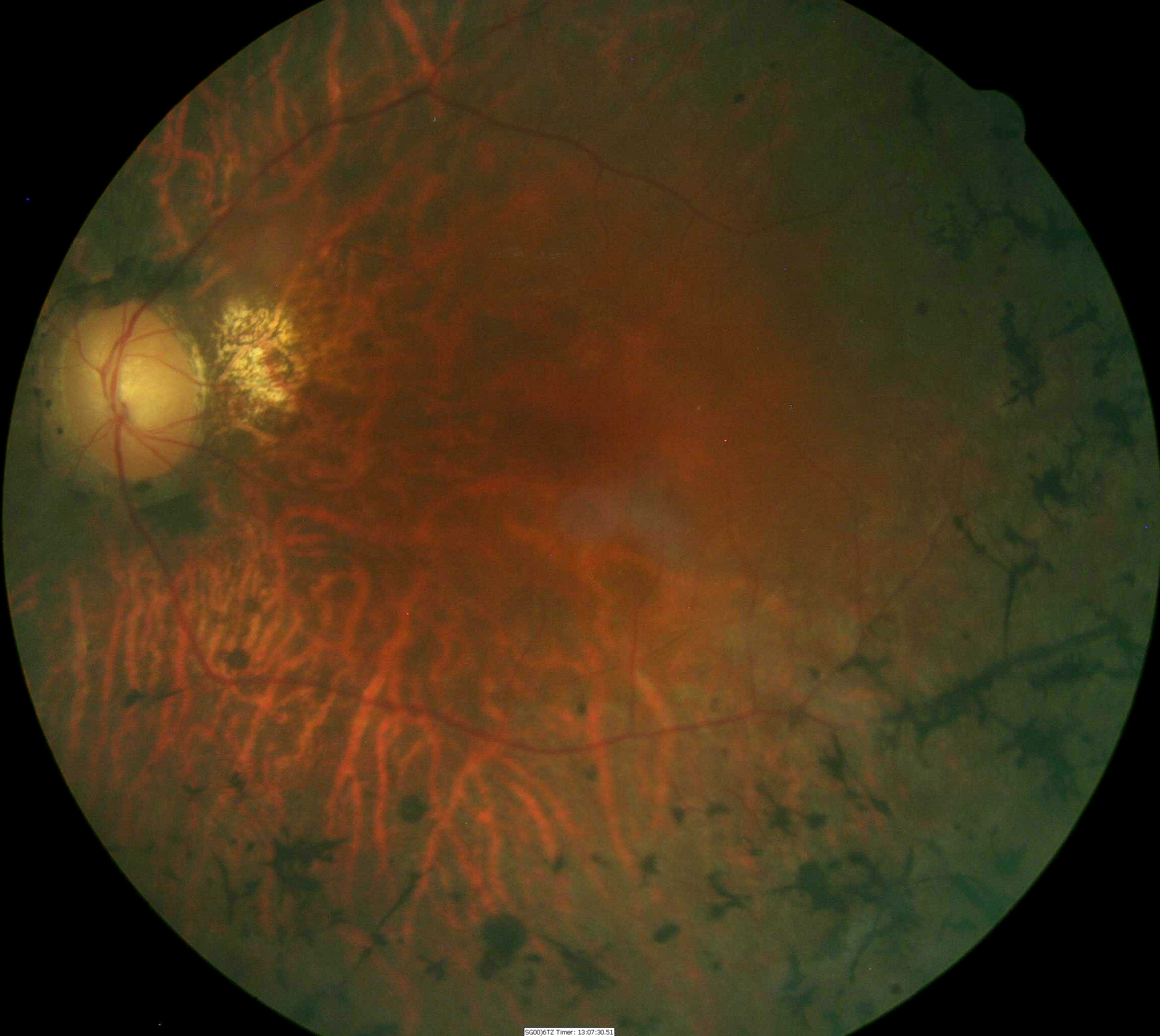
Fundus Appearance in Advanced Retinitis Pigmentosa
|
Retinitis pigmentosa is a rod-cone dystrophy that results in cell death, predominately in the rod photoreceptors. The genetic defects that cause retinitis pigmentosa can also affect the cone photoreceptors and the retinal pigment epithelium.
Structural Damage to the Eye
- The initial histopathologic change is a shortening of the outer rod segments
- The outer segments progressively shorten
- Rod photoreceptor dies, primarily in the retinal mid-periphery
- Cone apoptosis occurs in the same manner as rod cell death
- Cone cell death can occur early or late in the natural history of the disease
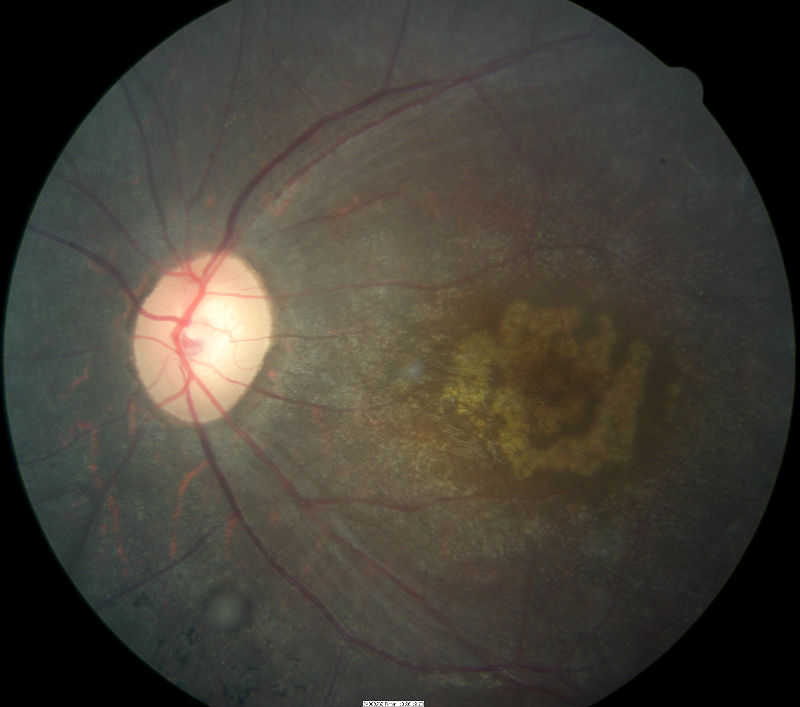
- Advanced cone dystrophy is characterized by central macular atrophy seen in the images below
 |
 |
Functional Damage to the Eye
- Loss of peripheral vision
- Loss of central acuity
The goal of a diagnostic evaluation in a patient with retiitis pigmentosa is to accomplish the following:
- Determine the correct diagnosis
- Offer genetic counseling
- Prescribe a treatment program to manage the disease and delay the loss of vision
Patient History
Patients with retinitis pigmentosa usually present with some or all of the following symptoms:
- Decreased vision at night
- Decreased peripheral vision
- Decreased central visual acuity
Ophthalmoscopy
Patient with retinitis pigmentosa usually present with some or all of the following clinical signs:
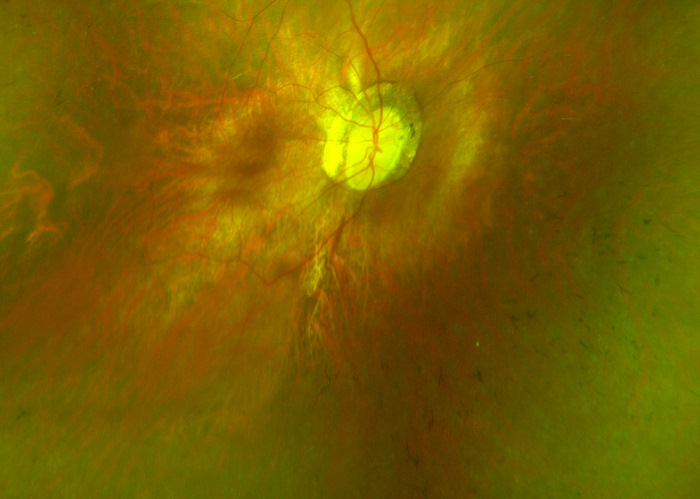
- Pigmentary degeneration in the retinal mid-periphery – “bone spicules”
- Wax-like pallor to the optic nerve
- Atrophy of the retinal pigment epithelium
- Arteriolar attenuation
- Central macular atrophy
Fundus images are from a 47-yer-old Hispanic male with retinitis pigmentosa.
- 20-year history of the disease
- 20/50 acuity in the right eye
- 20/60 acuity in the left eye
|
 |
DIAGNOSTIC TESTS
Electroretinography
- Electroretinogram testing evaluates the integrity of the retina
- Specialized pattern electroretinogram (PERG) test uses a contrast-reversing pattern for the stimulus to produce information about ganglion cell function
Visual Field Examination
- Automated threshold perimeters measure the visual field by plotting the threshold luminance value of the patient in various locations in the visual field
- The luminance of the light stimulus is represented by non-specific units of measurement called decibels (dB)
 |
Visual Field — Left Eye
|
Retinal Laser Scan — Macula
- The procedure can be accomplished by using the Cirrus OCT manufactured by Carl Zeiss Meditec
- Severe loss of retinal thickness at both maculas (e.g., central macular atrophy)
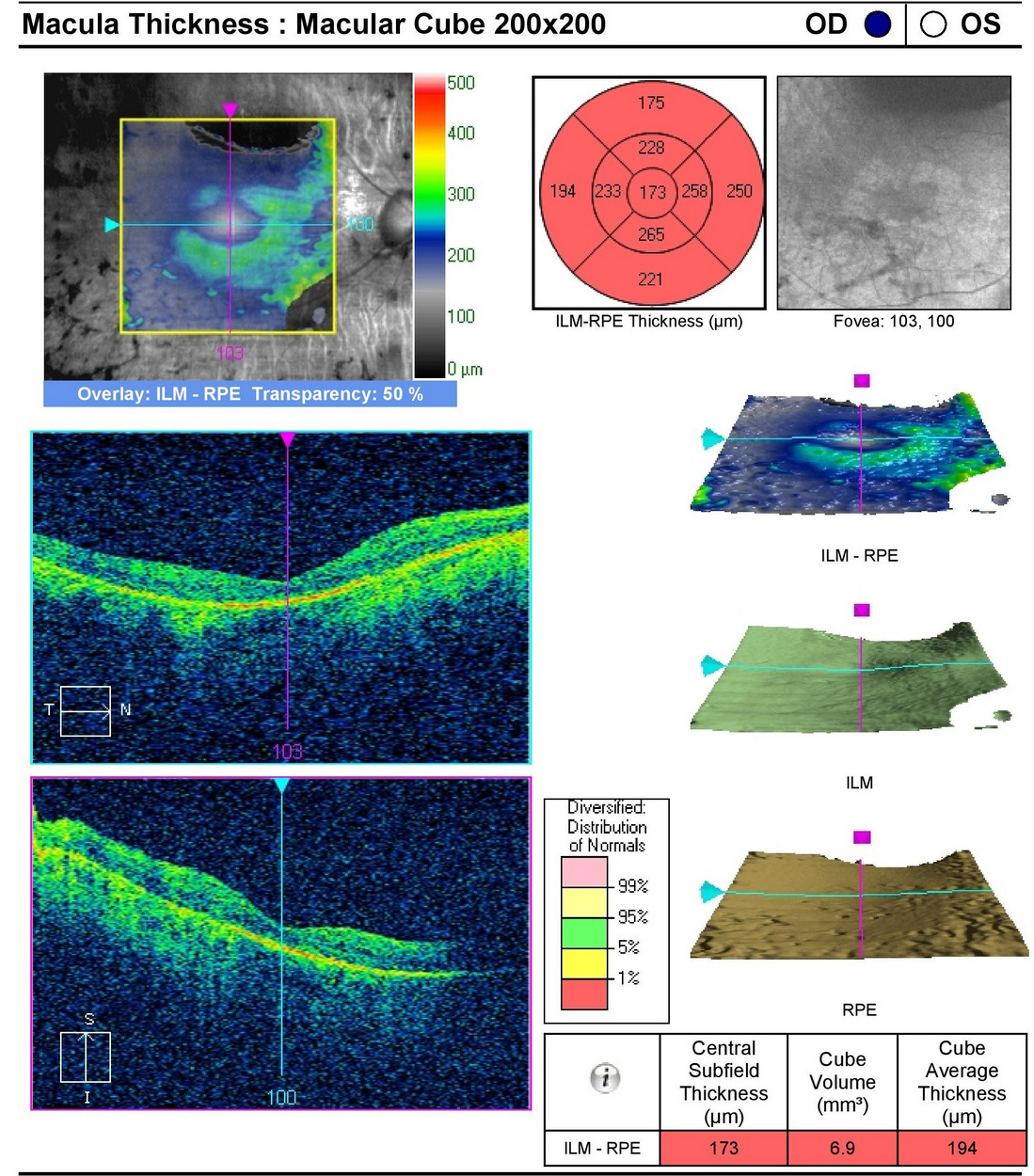 |
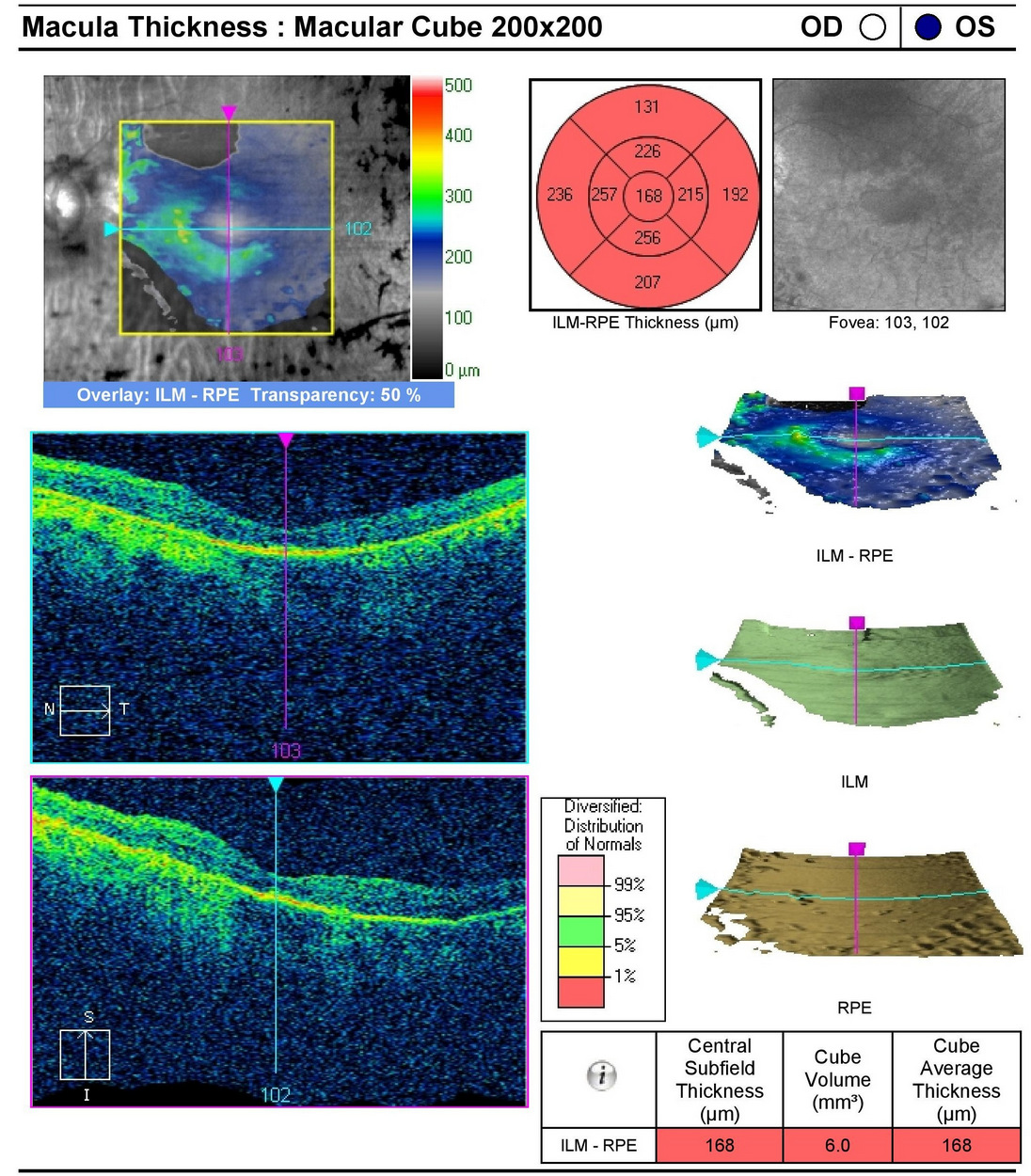 |
At present, there is no uniformly accepted classification system of retinitis pigmentosa.
The following diseases have clinical signs that can be similar to retinitis pigmentosa:
- Age-related macular degeneration, nonexudative
- Best’s disease
- Chloroquine/hydroxychloroquine toxicity
- Chorioretinopathy, central serous
- Chronic progressive external ophthalmoplegia
- Neuroretinitis, diffuse unilateral subacute
- Retinoschisis, juvenile
Medical Treatment
- Vitamin A/beta-carotene
- Docosahexaenoic acid (DHA)
- Acetazolamide
- Calcium channel blockers
- Lutein/zeaxanthin
- Valproic acid
Surgical Treatment
- Cataract extraction
- Growth factors
- Transplantation
- Retinal prosthesis
- Gene therapy
Other Forms of Treatment
- Consultations
- Diet
- Physical activity
1. Retinitis PIgmentosa. Handbook of Ocular Disease Management. http://www.reviewofoptometry.com/cmsdocuments/2012/6/ro0612_hndbk_em.pdf. Last accessed August 18, 2014.
2. Hope For RP Patients. RevOptom. 15 Mar 2012. http://www.revoptom.com/content/c/33121/. Last accessed August 18, 2014.
3. Telander D. Retinitis Pigmentosa. Medscape/EMedicine. 20 Feb 2014. http://emedicine.medscape.com/article/1227488-overview. Last accessed August 18, 2014.
362.74
Pigmentary retinal dystrophy
92083
Visual field examination
92250
Fundus photography
92283
Color vision examination
92275
Electroretinography
92225
Extended ophthalmoscopy
92015
Refraction
Occurrence
- The prevalence of typical RP is reported to be approximately 1 in 4000 in the United States
- The carrier state is believed to be approximately 1 in 100
- The highest reported frequency of occurrence for RP is among the Navajo Indians at 1 in 1878
Distribution
Generally, RP is distributed throughout the population evenly. However, because of X-linked factors that are only expressed in males, men may be slightly more affected than women.
- RP usually is diagnosed in young adulthood, although it can present anywhere from infancy to the mid-30’s to 50’s